What a Missed Recipe Parameter Actually Costs: The Deviation Cascade from Station to Station

Walk into a compounding suite mid-batch, and what you tend to find is not a breakdown at a single point, like a wrong setting, a missing entry, or a parameter no one confirmed, but the downstream consequence of one. The deviation report, when it comes, will trace back through two or three stations to locate the original error, and by the time it does, the batch has already been processed against the wrong value at every step that followed.
That is the pattern the industry calls a cascade, and it is more common than deviation metrics tend to suggest.
A single parameter verification failure at the start of a production sequence propagates downstream across every station that received the output of the one where the error first occurred. The QA investigation does not open at Station 1. It opens when someone at Station 3, or during in-process testing, sees a result that does not match expectations. At that point, the cost of the error is no longer the cost of the error; it is the cost of everything that has already happened since.

Deviation Reduction Programmes Address Outcomes. The Cascade Addresses Timing.
Deviation reduction programmes in pharma and biotech manufacturing typically focus on three levers: process tightening, training reinforcement, and documentation controls. All three matter, and none of them is wrong. But they are diagnostic responses to outcomes rather than to causes. The regulator’s own inspection record reflects this: procedures not followed and data integrity gaps are consistently among the most cited FDA Form 483 observations, and the standard response is to retrain the operator, when inspection analysts increasingly treat the same findings as a systemic condition the site created rather than a choice the individual made.
The parameter miss that starts a cascade is not usually a training failure or a documentation failure. It is a timing failure, a moment at which the operator at Station 1, needing to confirm a recipe value, did not have the EBR open in front of them. Either the MES terminal was at the other end of the room, the shared workstation was already in use, or the operator was working from a paper batch sheet that had not been updated to the current revision.
The miss was not deliberate and it was not due to a lack of understanding. The verification did not happen because the access point for the live record was somewhere other than where the operator was standing.
The Cascade Starts Before Anyone Knows There Is a Problem
The mechanics of a deviation cascade are not complicated, but the cost structure is.
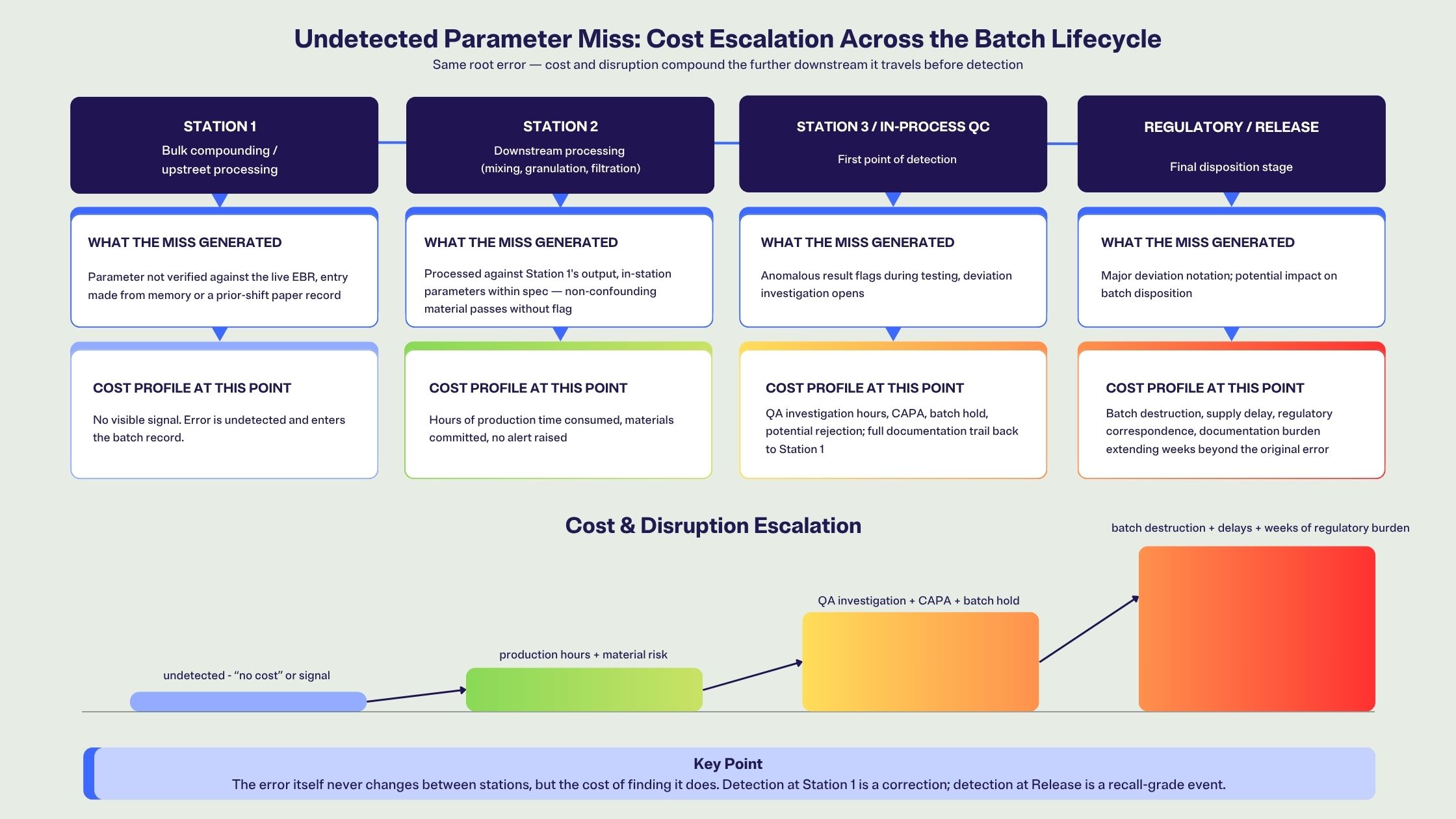
A recipe parameter error at Station 1 may not produce an immediately visible signal. The material might pass a visual check, and so the batch progresses. At Station 2, a downstream mixing or granulation step processes against what it received from Station 1. The process parameters for Station 2 are correct in isolation, but the material entering the step was already outside specification, and the Station 2 record will show a process that ran within its own parameters.
It surfaces at Station 3, or at in-process QC, or during finished product testing. When it does, the investigation has to reconstruct the sequence backwards: trace the Station 3 anomaly to the Station 2 inputs, trace those inputs to the Station 1 recipe verification, and then determine whether the verification actually took place against the current recipe or against an older revision, a printed record, or an operator's recall of the previous batch.
In most facilities, that reconstruction takes time, and by the time it is complete, the regulatory picture has also changed: a deviation that might have been classified as minor at the point of occurrence becomes major because of how far it travelled and what it carried with it.
The cost model is staged accordingly. According to data presented at PDA Week 2026 by Susan J. Schniepp and reported in Pharmaceutical Technology, the financial impact of a pharmaceutical quality failure depends almost entirely on when it is detected: a defect caught at incoming inspection remains minimal in cost, but a defect that reaches production can run to roughly $50,000, and one that reaches final product or triggers a recall can exceed $1 million. The cascade structure means the cost is always measured at the downstream discovery point, always after more work has been done against the same erroneous starting value. In biotech, the raw material and resource cost of a single lost batch is often put at $1 to $2 million, a figure attributed to Beth Junker, then senior director of bioprocess R&D at Merck, in BioProcess International.
The deviation report opens at the point of discovery. The cascade begins the moment a recipe parameter is not verified against the live record at the station where it matters.
What It Looks Like in Practice
Consider a fill-finish environment where an operator begins a batch at a compounding station.
- The MES terminal, the access point for the electronic batch record, the live recipe, and the current specification, is fixed to the wall at the suite entrance, seven metres from the compounding vessel. The operator has worked this suite for two years and knows the recipe well; the standard values have been consistent across recent batches. The current batch is a slight reformulation: a buffer concentration has changed, and the change is reflected in the EBR but not on the paper batch sheet that was printed from the previous revision.
- The operator confirms the parameter from the sheet; the value entered is the previous revision's value, not the current one, and the batch continues.
- At the downstream filling station, the in-process check for fill volume is within tolerance, and the filling line parameters are correct. The non-conforming material passes through two more operations before an out-of-specification result at QC flags it.
- The deviation investigation takes six working days, and the batch is rejected.
Place a GMP-rated mobile workstation at the compounding vessel, with the EBR live on-screen, updated to the current revision, so that the operator sees the changed parameter before making the entry. Whether that access point is a mobile workstation or a fixed in-room display positioned at the vessel matters less than the proximity itself: the record is where the work is. The verification happens at the moment of execution, against the current record, from the position where the work is taking place. The cascade does not start because the timing failure does not occur.
For Ops Managers, QA, and Manufacturing Leadership: The Questions Worth Asking
For operations managers and QA leads, the deviation cascade is a familiar pattern that tends to produce the same corrective response: a new procedure, a retraining event, or a SOP revision. Those interventions address the behaviour without addressing the condition that allowed the behaviour. The question worth asking is not why the operator did not verify the parameter but whether they had the access to do so, at the right moment, from where they were standing.
Practical questions for teams reviewing their deviation patterns:
For manufacturing leadership, the business case sits in the detection-stage cost model. Catching a parameter miss at Station 1, before the batch progresses, costs nothing beyond a correction. Catching it at Station 3 costs an investigation. Catching it at QC release costs a batch.
BioProcess International data puts the average biotech facility at one batch failure every 40.6 weeks. That number has since improved. By 2022, the same survey series (BioPlan Associates) reported the interval had stretched to roughly one failure every 58 weeks, a gain the analysts credit to hardware changes like single-use and continuous processing. Their read on what drives the next improvement is the useful part here: with the equipment problems largely addressed, the batch failures that remain trace back to the people and execution layer, which is exactly where point-of-work verification operates.
Point-of-Work Access Is the Infrastructure Layer That Closes the Verification Gap
Mobile and fixed workstations for life sciences manufacturing, GMP-rated, available across Grade C/D and Grade B environments, with stainless steel construction and MES-agnostic deployment, bring the EBR and live recipe to the station rather than requiring the operator to come to the terminal.
The ID-Flow 5 and ID-Flow 6 are the mobile platforms for Grade C/D and Grade B/C fill-finish, compounding, and packaging environments, with hot-swap battery architecture for continuous deployment across shifts without downtime for charging. The ID-Flow 9 and ID-View provide access in Grade B suites and critical zones where a mobile platform is not the appropriate form factor. In each case, the objective is the same: point-of-work visibility positioned at the station where the parameter is being applied, at the moment it is being applied.
Reducing deviations in life sciences manufacturing is not solely about improving processes or tightening controls. Compliance is managed by design and by operation: a process can be validated correctly and still fail at the point of execution if the operator cannot see the current record from where they are standing. The parameter miss that starts a cascade is a timing failure, and timing failures are infrastructure problems.
A Cascade Is a Record of a Gap, Not a Lapse
A deviation report that traces back through three stations to a parameter miss at the first is not, ultimately, a record of what an operator did wrong. It is a record of the gap between where the work happened and where the information needed to do it correctly was located. Close that gap by bringing the live EBR and current recipe to the station rather than expecting the operator to come to the terminal, and the cascade will not start. The cost, instead of compounding from station to station, does not occur.
If recipe verification is part of your deviation pattern, here's how our workstations can help
Kinetic ID builds GMP-rated mobile and fixed workstations for life sciences manufacturing environments, from Grade D processing areas through to Grade B fill-finish suites. The ID-Flow range covers mobile deployments with hot-swap battery architecture and validated stainless steel construction; the ID-View provides fixed in-room HMI for critical access points. Our solutions team can walk you through the product options, grade requirements, and deployment configurations relevant to your production stations and deviation reduction goals.
Speak with a solutions consultant
Frequently Asked Questions
References
- Schniepp, S.J. "Why Robust Quality Systems Save Pharmaceutical Companies Millions." Pharmaceutical Technology, PDA Week 2026. https://www.pharmtech.com/view/why-robust-quality-systems-save-pharmaceutical-companies-millions
- Biotech Facilities Average a Batch Failure Every 40.6 Weeks. BioProcess International. https://www.bioprocessintl.com/bioanalytical-methods/biotech-facilities-average-a-batch-failure-every-40-6-weeks
- FDA. Responding to FDA Form 483 Observations at the Conclusion of a Drug CGMP Inspection (draft guidance, March 2026). https://www.alston.com/en/insights/publications/2026/03/fda-guidance-drug-manufacturing-483-responses
- FDA 483 Observations FY 2024: Key Regulatory Issues. Compliance Insight. https://www.compliance-insight.com/fda-483-observations-fy-2024/
- Langer, E.S. Bioprocessing Sees Continued Improvements in Batch Failure Reductions in 2022. BioProcess Online (BioPlan Associates annual survey). https://www.bioprocessonline.com/doc/bioprocessing-sees-continued-improvements-in-batch-failure-reductions-in-0001
Calculate your productivity gain
Enter your site's operator headcount, shift pattern, and deviation rate, and get a payback estimate built on your numbers.
Try the ROI calculator